Journal of Marine Sciences ›› 2025, Vol. 43 ›› Issue (1): 22-33.DOI: 10.3969/j.issn.1001-909X.2025.01.003
Previous Articles Next Articles
Utilizing HCR-FISH to investigate the status of anaerobic methanotrophic archaea in cold seep sediments
HE Maoyu( ), WANG Jing, LI Sihan, LIANG Lewen*()
), WANG Jing, LI Sihan, LIANG Lewen*()
- School of Oceanography, Shanghai Jiao Tong University, Shanghai 200240, China
-
Received:2024-03-22Revised:2024-04-28Online:2025-03-15Published:2025-05-30 -
Contact:LIANG Lewen
CLC Number:
Cite this article
HE Maoyu, WANG Jing, LI Sihan, LIANG Lewen. Utilizing HCR-FISH to investigate the status of anaerobic methanotrophic archaea in cold seep sediments[J]. Journal of Marine Sciences, 2025, 43(1): 22-33.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: http://hyxyj.sio.org.cn/EN/10.3969/j.issn.1001-909X.2025.01.003
| 样品 编号 | 沉积物描述 | 沉积物柱状 样深度/cm | 样品数 量/个 |
|---|---|---|---|
| D231-1 | 还原性沉积物外部对照 | 14 | 7 |
| D231-2 | 还原性沉积物上边缘交界 | 30 | 15 |
| D231-3 | 还原性沉积物中心1 | 20 | 10 |
| D231-4 | 还原性沉积物中心2 | 18 | 9 |
| D231-5 | 还原性沉积物中心下边缘交界 | 22 | 11 |
| D231-6 | 还原性沉积物白色菌席 | 18 | 9 |
| 合计 | 61 | ||
Tab.1 Information of sediment samples in Formosa cold seep used in this study
| 样品 编号 | 沉积物描述 | 沉积物柱状 样深度/cm | 样品数 量/个 |
|---|---|---|---|
| D231-1 | 还原性沉积物外部对照 | 14 | 7 |
| D231-2 | 还原性沉积物上边缘交界 | 30 | 15 |
| D231-3 | 还原性沉积物中心1 | 20 | 10 |
| D231-4 | 还原性沉积物中心2 | 18 | 9 |
| D231-5 | 还原性沉积物中心下边缘交界 | 22 | 11 |
| D231-6 | 还原性沉积物白色菌席 | 18 | 9 |
| 合计 | 61 | ||
| 探针名称 | 靶向类群 | 探针序列(5’-3’) | 来源 | |
|---|---|---|---|---|
| 起始 探针 | ARCH915-H | Archaea | CCGAATACAAAGCATCAACGACTAGAAAAAAGTGCTCCCCCGCCAATTCCT | 文献[ |
| EUB338-R | Bacteria | TACGCCCTAAGAATCCGAACCCTATGAAATAGCTGCCTCCCGTAGGAGT | 文献[ | |
| ANME-1-350-H | ANME-1 | CCGAATACAAAGCATCAACGACTAGAAAAAAAGTTTTCGCGCCTGATGC | 文献[ | |
| ANME-2-538-H | ANME-2 | CCGAATACAAAGCATCAACGACTAGAAAAAAGGCTACCACTCGGGCCGC | 本研究 | |
| ANME-3-1249-H | ANME-3 | CCGAATACAAAGCATCAACGACTAGAAAAAATCGGAGTAGGGACCCATT | 本研究 | |
| DSS658-R | Desulfosarcina-Desulfococcus | TACGCCCTAAGAATCCGAACCCTATGAAATATCCACTTCCCTCTCCCAT | 本研究 | |
| 扩增 探针 | H1 | CATAGGGTTCGGATTCTTAGGGCGTAGCAGCATCAATACGCCCTAAGAATCC | 文献[ | |
| H2 | TACGCCCTAAGAATCCGAACCCTATGGGATTCTTAGGGCGTATTGATGCTGC | 文献[ | ||
| R1 | TCTAGTCGTTGATGCTTTGTATTCGGCGACAGATAACCGAATACAAAGCATC | 文献[ | ||
| R2 | CCGAATACAAAGCATCAACGACTAGAGATGCTTTGTATTCGGTTATCTGTCG | 文献[ | ||
Tab.2 HCR-FISH probes used in this study
| 探针名称 | 靶向类群 | 探针序列(5’-3’) | 来源 | |
|---|---|---|---|---|
| 起始 探针 | ARCH915-H | Archaea | CCGAATACAAAGCATCAACGACTAGAAAAAAGTGCTCCCCCGCCAATTCCT | 文献[ |
| EUB338-R | Bacteria | TACGCCCTAAGAATCCGAACCCTATGAAATAGCTGCCTCCCGTAGGAGT | 文献[ | |
| ANME-1-350-H | ANME-1 | CCGAATACAAAGCATCAACGACTAGAAAAAAAGTTTTCGCGCCTGATGC | 文献[ | |
| ANME-2-538-H | ANME-2 | CCGAATACAAAGCATCAACGACTAGAAAAAAGGCTACCACTCGGGCCGC | 本研究 | |
| ANME-3-1249-H | ANME-3 | CCGAATACAAAGCATCAACGACTAGAAAAAATCGGAGTAGGGACCCATT | 本研究 | |
| DSS658-R | Desulfosarcina-Desulfococcus | TACGCCCTAAGAATCCGAACCCTATGAAATATCCACTTCCCTCTCCCAT | 本研究 | |
| 扩增 探针 | H1 | CATAGGGTTCGGATTCTTAGGGCGTAGCAGCATCAATACGCCCTAAGAATCC | 文献[ | |
| H2 | TACGCCCTAAGAATCCGAACCCTATGGGATTCTTAGGGCGTATTGATGCTGC | 文献[ | ||
| R1 | TCTAGTCGTTGATGCTTTGTATTCGGCGACAGATAACCGAATACAAAGCATC | 文献[ | ||
| R2 | CCGAATACAAAGCATCAACGACTAGAGATGCTTTGTATTCGGTTATCTGTCG | 文献[ | ||
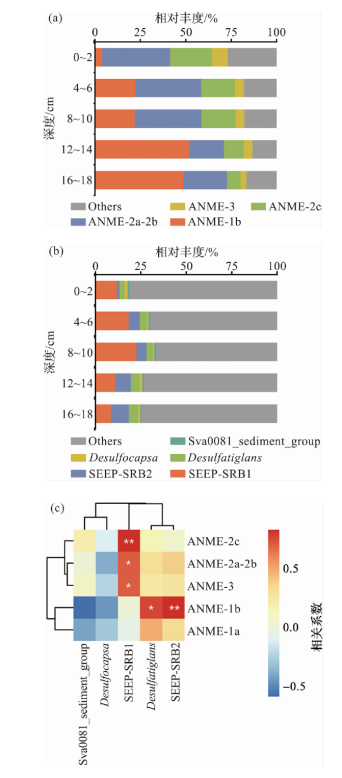
Fig.1 Composition and correlation of ANME and SRB groups in the sediment samples of Formosa cold seep (Figure a illustrates the variation of ANME groups with depth in site D231-4, where “Others” in the figure represents archaea other than ANME and ANME with ASV abundances less than 1%. Figure b depicts the variation of SRB groups with depth in site D231-4, where “Others” denotes bacteria other than SRB and SRB with ASV abundances less than 1%. Figure c presents a Pearson correlation heatmap of ANME and SRB groups across all depth intervals from station D231, based on 61 sediment samples. In the heatmap, * indicates p<0.05, and ** indicates p<0.01.)
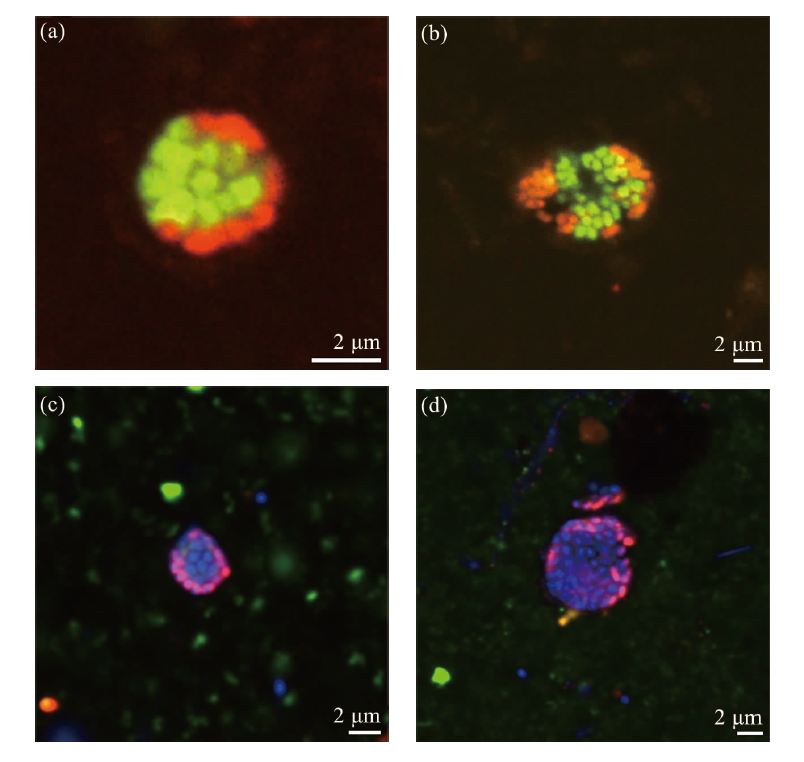
Fig.2 HCR-FISH images of ANME-SRB consortia in the sediment samples of Formosa cold seep (Figure a illustrates microbial consortia composed of archaea and bacteria. Figure b displays ANME-SRB consortia consisting of ANME-2 and sulfate-reducing bacteria (SRB). No hybridization signals were observed for ANME-SRB consortia that were positively hybridized with the DSS probe when probed with ANME-1-targeting (Figure c) or ANME-3-targeting (Figure d) probes. Oligonucleotide probes DSS658-R and EUB338-R were employed to target SRB and bacteria, respectively, and were labeled red. Probes ARCH915-H, ANME-1-350-H, ANME-2-538-H, and ANME-3-1249-H were used to target archaea, ANME-1, ANME-2, and ANME-3, respectively, and were labeled green. DAPI staining was applied for nuclear staining and appeared blue.)
| 层位 | 细胞团数量/个 | ||
|---|---|---|---|
| ANME-1 | ANME-2 | ANME-3 | |
| 0~2 cm | 0 | 86 | 0 |
| 4~6 cm | 0 | 71 | 0 |
| 8~10 cm | 0 | 73 | 0 |
| 12~14 cm | 0 | 82 | 0 |
| 16~18 cm | 0 | 78 | 0 |
| 平均 | 0 | 78 | 0 |
Tab.3 The ANME consortia number observed in the sediments at different depths
| 层位 | 细胞团数量/个 | ||
|---|---|---|---|
| ANME-1 | ANME-2 | ANME-3 | |
| 0~2 cm | 0 | 86 | 0 |
| 4~6 cm | 0 | 71 | 0 |
| 8~10 cm | 0 | 73 | 0 |
| 12~14 cm | 0 | 82 | 0 |
| 16~18 cm | 0 | 78 | 0 |
| 平均 | 0 | 78 | 0 |
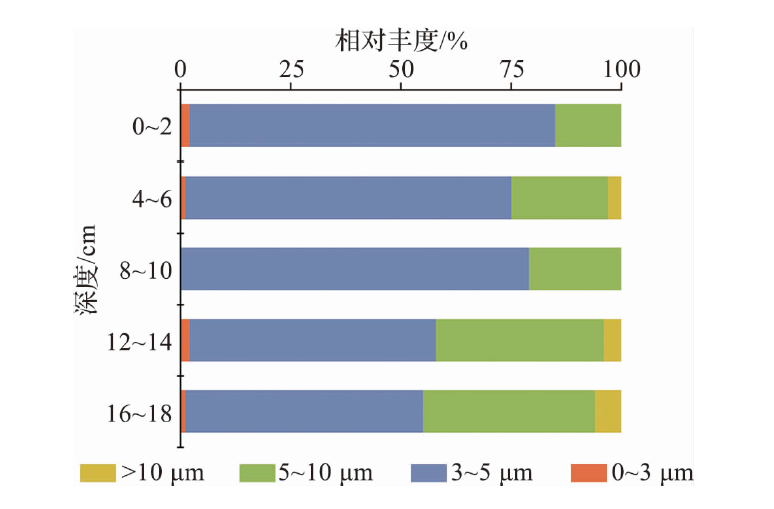
Fig.3 The composition of the consortia size in the sediment samples of Formosa cold seep
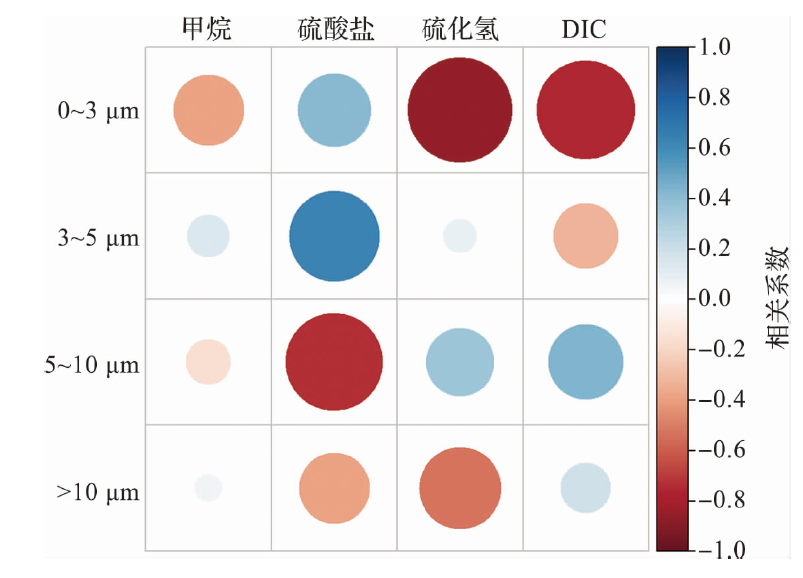
Fig.4 Heatmap of Pearson’s correlation coefficients between environmental factors and consortia sizes
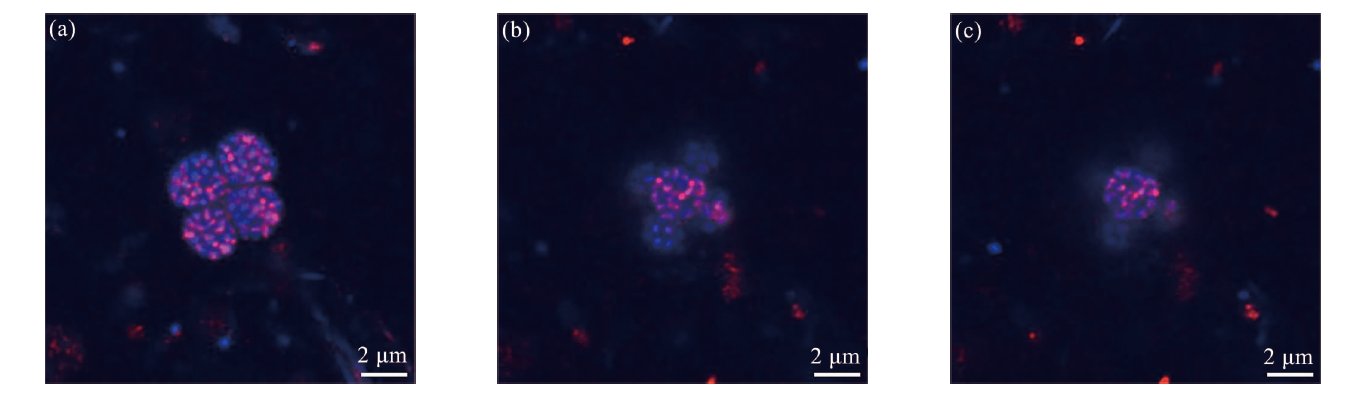
Fig.5 The HCR-FISH images of the consortia cluster (The microbial consortia exhibited positive hybridization with the SRB-targeting probe (DSS658-R, red), DAPI staining appeared blue. Figures a to c represent scanning results from different z-axes.)
| [1] |
KNITTEL K, BOETIUS A. Anaerobic oxidation of methane: Progress with an unknown process[J]. Annual Review of Microbiology, 2009, 63: 311-334.
DOI PMID |
| [2] |
PAULL C K, HECKER B, COMMEAU R, et al. Biological communities at the Florida escarpment resemble hydrothermal vent taxa[J]. Science, 1984, 226(4677): 965-967.
PMID |
| [3] | FENG D, QIU J W, HU Y, et al. Cold seep systems in the South China Sea: An overview[J]. Journal of Asian Earth Sciences, 2018, 168: 3-16. |
| [4] | SCHREIBER L, HOLLER T, KNITTEL K, et al. Identi-fication of the dominant sulfate-reducing bacterial partner of anaerobic methanotrophs of the ANME-2 clade[J]. Environmental Microbiology, 2010, 12(8): 2327-2340. |
| [5] | SCHÖNHUBER W, FUCHS B, JURETSCHKO S, et al. Improved sensitivity of whole-cell hybridization by the combination of horseradish peroxidase-labeled oligonucleotides and tyramide signal amplification[J]. Applied and Environ-mental Microbiology, 1997, 63(8): 3268-3273. |
| [6] | VOLPI E V, BRIDGER J M. FISH glossary: An overview of the fluorescence in situ hybridization technique[J]. Bio-Techniques, 2008, 45(4): 385-409. |
| [7] |
AMANN R, FUCHS B M. Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques[J]. Nature Reviews Microbiology, 2008, 6(5): 339-348.
DOI PMID |
| [8] |
YAMAGUCHI T, KAWAKAMI S, HATAMOTO M, et al. In situ DNA-hybridization chain reaction (HCR): A facilitated in situ HCR system for the detection of environmental microorganisms[J]. Environmental Microbiology, 2015, 17(7): 2532-2541.
DOI PMID |
| [9] |
YAMAGUCHI T, FUCHS B M, AMANN R, et al. Rapid and sensitive identification of marine bacteria by an improved in situ DNA hybridization chain reaction (quickHCR-FISH)[J]. Systematic and Applied Microbiology, 2015, 38(6): 400-405.
DOI PMID |
| [10] | BHATTARAI S, CASSARINI C, LENS P L. Physiology and distribution of archaeal methanotrophs that couple anaerobic oxidation of methane with sulfate reduction[J]. Microbiology and Molecular Biology Reviews, 2019, 83(3): e00074-18. |
| [11] |
KNITTEL K, LÖSEKANN T, BOETIUS A, et al. Diversity and distribution of methanotrophic Archaea at cold seeps[J]. Applied and Environmental Microbiology, 2005, 71(1): 467-479.
PMID |
| [12] | NAUHAUS K, ALBRECHT M, ELVERT M, et al. In vitro cell growth of marine archaeal-bacterial consortia during anaerobic oxidation of methane with sulfate[J]. Environ-mental Microbiology, 2007, 9(1): 187-196. |
| [13] | NIU M Y, FAN X B, ZHUANG G C, et al. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea[J]. FEMS Microbiology Ecology, 2017, 93(9): 105-110. |
| [14] |
RUFF S E, BIDDLE J F, TESKE A P, et al. Global dispersion and local diversification of the methane seep microbiome[J]. Proceedings of the National Academy of Sciences of the United States of America, 2015, 112(13): 4015-4020.
DOI PMID |
| [15] | 孙瑜, 牛明杨, 刘俏, 等. 南海Formosa冷泉区沉积物微生物多样性与分布规律研究[J]. 微生物学报, 2022, 62(6):2001-2020. |
| SUN Y, NIU M Y, LIU Q, et al. Diversity and distribution of microorganisms in the sediment of Formosa cold seep in South China Sea[J]. Acta Microbiologica Sinica, 2022, 62(6): 2001-2020. | |
| [16] | JIA Z Y, DONG Y J, XU H, et al. Optimizing the hybridization chain reaction-fluorescence in situ hybridization (HCR-FISH) protocol for detection of microbes in sediments[J]. Marine Life Science & Technology, 2021, 3(4): 529-541. |
| [17] | BOLYEN E, RIDEOUT J R, DILLON M R, et al. Repro-ducible, interactive, scalable and extensible microbiome data science using QIIME 2[J]. Nature Biotechnology, 2019, 37(8): 852-857. |
| [18] |
QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools[J]. Nucleic Acids Research, 2013, 41(Database issue): 590-596.
DOI PMID |
| [19] | WEI T, SIMKO V, LEVY M, et al. corrplot: Visualization of a correlation matrix[CP]. (2017-10-16). https://CRAN.R-project.org/package=corrplot. |
| [20] | KOLDE R. pheatmap: Pretty Heatmaps[CP]. (2022-10-14). https://CRAN.R-project.org/package=pheatmap. |
| [21] |
CHOI H M T, CHANG J Y, TRINH L A, et al. Programmable in situ amplification for multiplexed imaging of mRNA expression[J]. Nature Biotechnology, 2010, 28(11): 1208-1212.
DOI PMID |
| [22] | MATSUBAYASHI M, SHIMADA Y, LI Y Y, et al. Phylogenetic diversity and in situ detection of eukaryotes in anaerobic sludge digesters[J]. PLoS One, 2017, 12(3): e0172888. |
| [23] |
MOTER A, GÖBEL U B. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms[J]. Journal of Microbiological Methods, 2000, 41(2): 85-112.
DOI PMID |
| [24] | LEVSKY J M, SINGER R H. Fluorescence in situ hybridization: Past, present and future[J]. Journal of Cell Science, 2003, 116(14): 2833-2838. |
| [25] | BOETIUS A, RAVENSCHLAG K, SCHUBERT C J, et al. A marine microbial consortium apparently mediating anaerobic oxidation of methane[J]. Nature, 2000, 407(6804): 623-626. |
| [26] |
PERNTHALER A, DEKAS A E, BROWN C T, et al. Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(19): 7052-7057.
DOI PMID |
| [27] | LASO-PÉREZ R, WU F B, CRÉMIÈRE A, et al. Evolutionary diversification of methanotrophic ANME-1 archaea and their expansive virome[J]. Nature Microbiology, 2023, 8(2): 231-245. |
| [28] |
MAIGNIEN L, PARKES R J, CRAGG B, et al. Anaerobic oxidation of methane in hypersaline cold seep sediments[J]. FEMS Microbiology Ecology, 2013, 83(1): 214-231.
DOI PMID |
| [29] |
STOKKE R, ROALKVAM I, LANZEN A, et al. Integrated metagenomic and metaproteomic analyses of an ANME-1-dominated community in marine cold seep sediments[J]. Environmental Microbiology, 2012, 14(5): 1333-1346.
DOI PMID |
| [30] |
KEVORKIAN R T, CALLAHAN S, WINSTEAD R, et al. ANME-1 archaea may drive methane accumulation and removal in estuarine sediments[J]. Environmental Microbiology Reports, 2021, 13(2): 185-194.
DOI PMID |
| [31] |
RISTOVA P P, WENZHÖFER F, RAMETTE A, et al. Spatial scales of bacterial community diversity at cold seeps (Eastern Mediterranean Sea)[J]. ISME Journal, 2015, 9(6): 1306-1318.
DOI PMID |
| [32] | CHEN Y, XU C L, WU N Y, et al. Diversity of anaerobic methane oxidizers in the cold seep sediments of the Okinawa trough[J]. Frontiers in Microbiology, 2022, 13: 819187. |
| [33] | MCGLYNN S E, CHADWICK G L, KEMPES C P, et al. Single cell activity reveals direct electron transfer in methanotrophic consortia[J]. Nature, 2015, 526(7574): 531-535. |
| [34] |
WANKEL S D, ADAMS M M, JOHNSTON D T, et al. Anaerobic methane oxidation in metalliferous hydrothermal sediments: Influence on carbon flux and decoupling from sulfate reduction[J]. Environmental Microbiology, 2012, 14(10): 2726-2740.
DOI PMID |
| [35] | MCILROY S J, LEU A O, ZHANG X Q, et al. Anaerobic methanotroph ‘Candidatus Methanoperedens nitroreducens’ has a pleomorphic life cycle[J]. Nature Microbiology, 2023, 8(2): 321-331. |
| [36] |
ORPHAN V J, HOUSE C H, HINRICHS K U, et al. Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(11): 7663-7668.
DOI PMID |
| [37] |
KRUKENBERG V, RIEDEL D, GRUBER-VODICKA H R, et al. Gene expression and ultrastructure of meso- and thermophilic methanotrophic consortia[J]. Environmental Microbiology, 2018, 20(5): 1651-1666.
DOI PMID |
| [38] |
KLEINDIENST S, RAMETTE A, AMANN R, et al. Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments[J]. Environmental Microbiology, 2012, 14(10): 2689-2710.
DOI PMID |
| [1] | HE Xinyi, LIU Qian, LI Xiaohu, LI Zhenggang, WANG Hao, ZHU Zhimin, LI Huaiming. Microbial community structure and function in deep-sea polymetallic nodules and surrounding sediments [J]. Journal of Marine Sciences, 2025, 43(1): 34-46. |
| [2] | WANG Tianyi, DONG Yanhui, CHU Fengyou, SHI Xuefa, LI Xiaohu, SU Rong, ZHANG Weiyan. Classification and genesis of deep-sea REY-rich sediments in the Pacific Ocean [J]. Journal of Marine Sciences, 2024, 42(1): 23-35. |
| [3] | ZHAO Xuekai, GUO Kaiyuan, ZHOU Yunhao, JIA Liyuan, YANG Zhibo, ZHANG Qinxu, ZHANG Mingliang. Spatio-temporal variation of suspended sediment and its dynamic factors in Liaohe Estuary [J]. Journal of Marine Sciences, 2024, 42(1): 36-46. |
| [4] | WU Xinran, DONG Yanhui, LI Zhenggang, WANG Hao, ZHANG Weiyan, LI Huaiming, LI Xiaohu, CHU Fengyou. Deep-sea rare earth resource potential in the Eastern Pacific Clarion-Clipperton Fracture Zone: Constraint from sediment geochemistry [J]. Journal of Marine Sciences, 2023, 41(4): 46-56. |
| [5] | WANG Ying, TAO Chunhui, ZHANG Guoyin, ZHOU Jianping, SHEN Honglei. Simulation study on oblique in situ acoustic longitudinal wave measurement of seafloor inhomogeneous sedimentary layer [J]. Journal of Marine Sciences, 2023, 41(4): 57-69. |
| [6] | REN Shijun, ZHANG Li, WANG Hongguang, ZHANG Qinghong, ZHUANG Tonghui, WEI Na, SONG Jihong, CHENG Luxian, WANG You, MU Qinglin. Distribution and interannual changes of PAHs in surface sediments from the adjacent waters of the green petrochemical project [J]. Journal of Marine Sciences, 2023, 41(3): 83-91. |
| [7] | ZHANG Wanying, LU Shasha, XIA Xiaoming, LIU Jingui. Variations in water and sediment fluxes in Oujiang River during flooding and non-flooding seasons [J]. Journal of Marine Sciences, 2023, 41(2): 61-70. |
| [8] | CHEN Yining, CHEN Luzhen. Interactions between vegetation and sediment carbon pools within coastal blue carbon ecosystems: A review and perspective [J]. Journal of Marine Sciences, 2023, 41(1): 3-13. |
| [9] | LIU Liping, CHU Fengyou, GUO Lei, LI Xiaohu. Explorations of marine gas hydrate deposits and the signatures of hydrocarbon venting using in situ techniques [J]. Journal of Marine Sciences, 2023, 41(1): 26-44. |
| [10] | WANG Lei, LI Chunfeng, LI Kedi, YAO Zewei, TAO Tiansheng. Characteristics and tectonic implications of the Mesozoic residual strata in the East China Sea Shelf Basin [J]. Journal of Marine Sciences, 2022, 40(4): 11-24. |
| [11] | DU Yichao, LUO Xiaowen, WANG Jun, CUI Jiaxin. Characteristics of runoff and sediment discharge in the Pearl River Basin in recent 70 years and analysis on the affecting factors of human activities [J]. Journal of Marine Sciences, 2022, 40(4): 52-64. |
| [12] | YAO Huabo, ZHANG Zhaohui, JIN Haiyan, CHEN Jianfang. Influencing factors and distribution of particulate phosphorus in the surface sediments of the Changjiang Estuary and coastal waters of Zhejiang [J]. Journal of Marine Sciences, 2022, 40(4): 73-81. |
| [13] | LIN Junchuan, KONG Deming, CHEN Fajin, HUANG Chao, . Climatic and environmental changes over the last 1 000 years as recorded by the sediments in Beibu Gulf [J]. Journal of Marine Sciences, 2022, 40(3): 49-61. |
| [14] | . Occurrences, sources and ecological risks of organophosphate esters: Case study for surface sediments in Beibu Gulf, South China Sea [J]. Journal of Marine Sciences, 2022, 40(3): 99-108. |
| [15] | LUAN Kuifeng, XU Hang, PAN Yujia, et al. Study on spectral curve characteristics of surface suspended sediment concentration in the Changjiang (Yangtze River) Estuary based on hyperspectral sensor#br# [J]. Journal of Marine Sciences, 2022, 40(1): 64-71. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||